其实这篇文章揭开一个很尴尬的事情,那就是,我们当前关于病毒的溯源完全受制于了早期提交的武汉本地病毒基因数据(偏倚性太高了)。
1,华南海鲜市场依然是最大的引爆点,超级传播病毒就是从这里出去的
2,有武汉旅行史的广东人和美国人感染了进化上更加靠前的病毒,而这个病毒却在武汉提交的数据里没发现,这说明当时武汉提交的数据实在是过于特殊,不具有普遍性,而理论上,可能当地存在更多的被感染者,他们更早接触了病毒,但是和华南海鲜市场没有直接交集。
3,加大对武汉当地的一些数据广泛性查找,可以更好地定位到相对更早的病例,也就是很多人一直追问的0号病人,只有找到这个人,或者这一些人,我们才能真正找到武汉病毒的源头,然后彻底掐断传染源。这一点武汉是有能力的,他们有样本,有强大的二代测序能力。
文章解读在后文,重点分析在前。
—————外地起源论?no,依然是武汉。—————
估计很多人看到题目,已经浮想联翩了,比如美国的和广东的竟然比华南海鲜市场的病毒还古老等等,但是我要说,别着急。
尽管从进化上分析,我们发现美国的和广东的病例说携带的病毒属于较为古老的病毒,但是,这并不能说明病毒是其他地方的,事实上,这两个患者,他们的旅行记录表明 2019 年 12 月底至 2020 年 1 月初都来过武汉,然后他们都是1月份发病的。所以,他们是在武汉被感染的。上面的研究,只能佐证他们并不是被华南海鲜市场病毒感染的,而是来自武汉其他地方的病毒。而这也引出了一个本研究的根本上缺陷——病毒样本不完善
广东首例:
They had no history of contact with animals, visits to markets including the Huanan seafood whole sale in Wuhan, or eating game meat in restaurants.——柳叶刀[1]
美国首例:
Although the patient reported that he had not spent time at the Huanan seafood market and reported no known contact with ill persons during his travel to China——新英格兰[2]
2。研究存在的缺陷
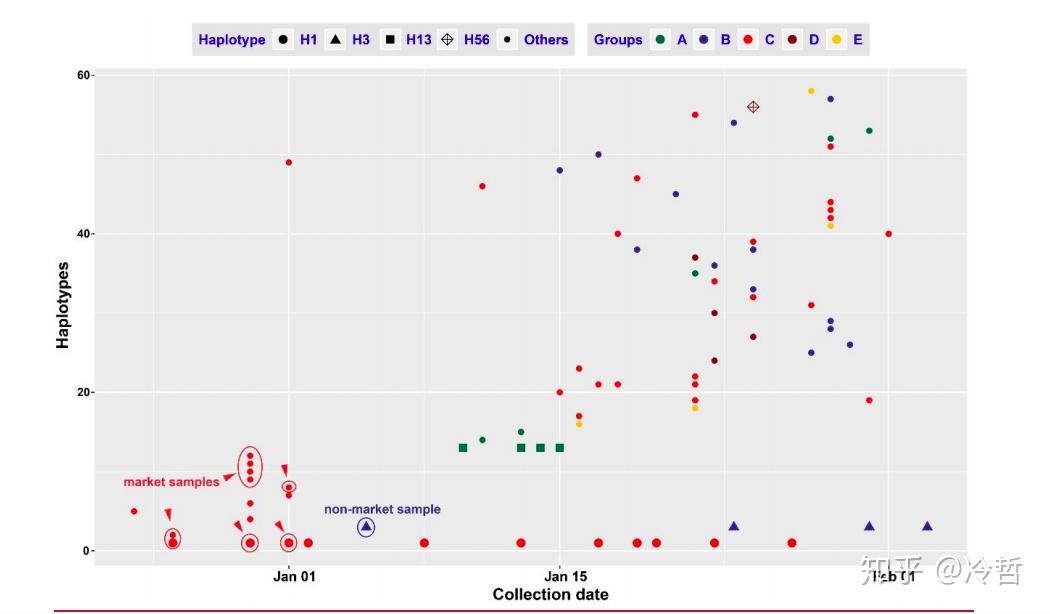
事实上,尽管整个研究选择了93个样本的基因组,是迄今为止发表的关于新型冠状病毒疫情最大规模群体基因组研究,但是,这个研究样本还是不够全面。武汉样本样品采集时间局限于 2019 年 12 月 24 日和 2020 年 1 月 5 日。而那个时候,当地对于疫情的排查集中在了华南海鲜市场,而样本来自几家定点医院,换句话说,样本取样地不具备统计学上的无偏性,这导致最终的数据分析也必然受到了影响,也就是garbage in,garbage out。事实上,我们根据广东和美国的病例,完全可以推测出,武汉本地存在更古老的单倍型H13 和 H38。因此,要想完整的溯源,事实上对武汉后续发病例进行更广泛的基因组检测,这样才能更好地确定真正的源头。
3. 即使不是华南海鲜市场,也依然是武汉或者湖北
基于上述分析,其实我们可以看到,尽管大概率排除了华南海鲜市场为首发地,但是也不尽然,因为可能华南海鲜市场存在一些病例自愈了而没有被排查到。但是,即便抛开这个小概率事件,整个病毒的溯源来看,这个病毒集中爆发地,依然是存在于武汉或者湖北。
下图是文章里提到的路线,
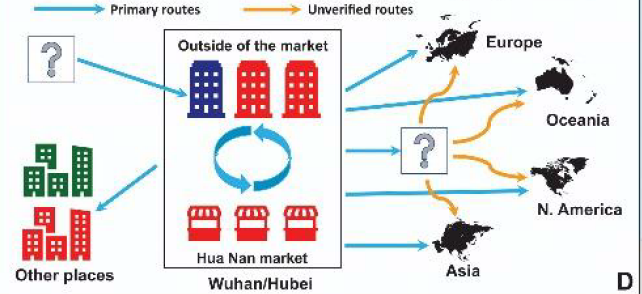
简单地说,某些未知来源的病毒出现在了武汉,不过这个病毒应该存在于华南海鲜市场和其他地方,然后不断扩散到全世界。
而到目前为止,所有的病例,源头都可以追溯到武汉上,所以,武汉必然是当前疫情的第一个出发点。
至于武汉的病毒从哪里来?我们不得而知。它们究竟是如何出现在武汉,也不得而知,所以我们需要更多的、更全面的数据,而这些,就要期待研究人员和科研机构来贡献了。
有两种猜测:
早期只是轻微传播,至少从目前来看,连祖先型的H13等都会感染人,那么,这个更古老的未知病毒,经历了一次重大的变异才忽然爆发出如此强大的杀伤力
2.早期的病毒是在宿主身上,它在武汉第一次感染了人
这个可能更靠谱一些,就是病毒可能一直在动物宿主身上并不断进化,然后在武汉的时候,这个病毒第一次传播到了人身上,然后从人传到了更多的人身上。
二,一些额外的期待
目前病毒已经发生了许多突变,下图是截至今天为止所有已经报道的病毒身上的突变,我们可以看到均匀的分布在全部基因组上[3]
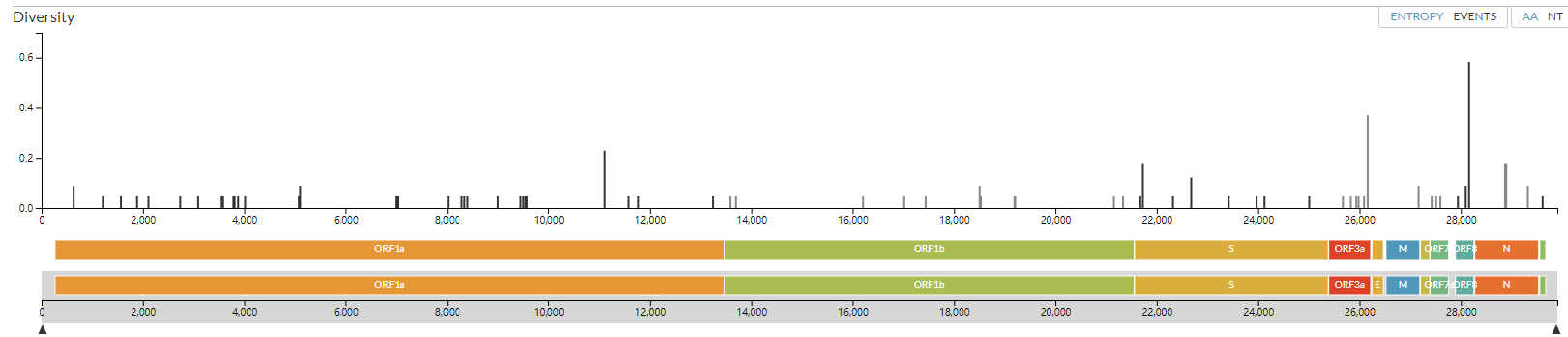
而这些突变,可能引发一些潜在的威胁。
以中科院这篇文章为例,他们研究的120个突变的核苷酸关联了119个氨基酸密码子,其中79个密码子 (65.83%)改变了氨基酸类型,并有42个(53.17%)氨基酸理化性质都被改变。这是不容小觑的,蛋白是生物体内发挥生命理化作用的主要分子,氨基酸的改变,往往会引发蛋白质本身理化性质的改变,而这些改变,是否会影响新型冠状病毒的传染性、致病性等问题,都是值得重视的。
—————原文解读部分———————
一、华南海鲜市场是不是真正的病毒源头?
2月19日,中国科学院西双版纳热带植物园的研究人员在预印网站上分享了一篇对新型冠状病毒肺炎的溯源研究,在该研究中,作者提出了一个看法:华南海鲜市场并不是病毒的源头[4]。
这篇研究是怎么做的呢?
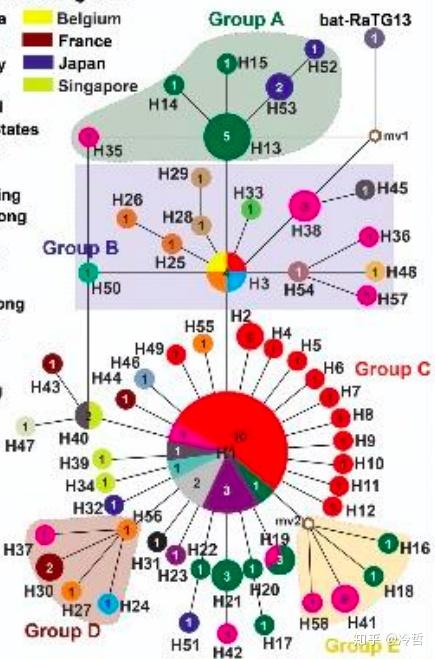
研究人员首先从公共数据库GISAID EpiFluTM里下载了来自亚、欧、澳和北美四个州12个国家的93个新型冠状病毒的全基因组数据,通过全基因组分析来进行溯源传染源和扩散路径的研究。依据其基因组上的变异位点对这些病毒进行分类,最后得出了58种单倍型,他们可以归纳成五大类,如下图所示
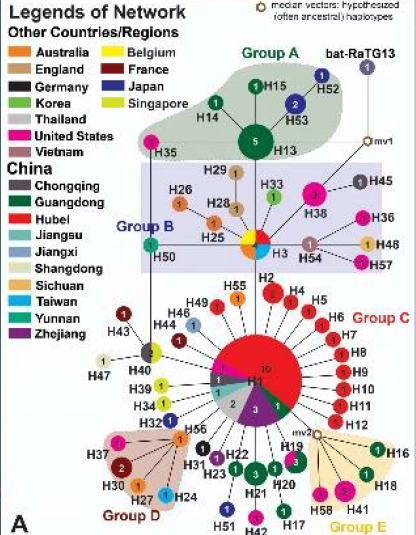
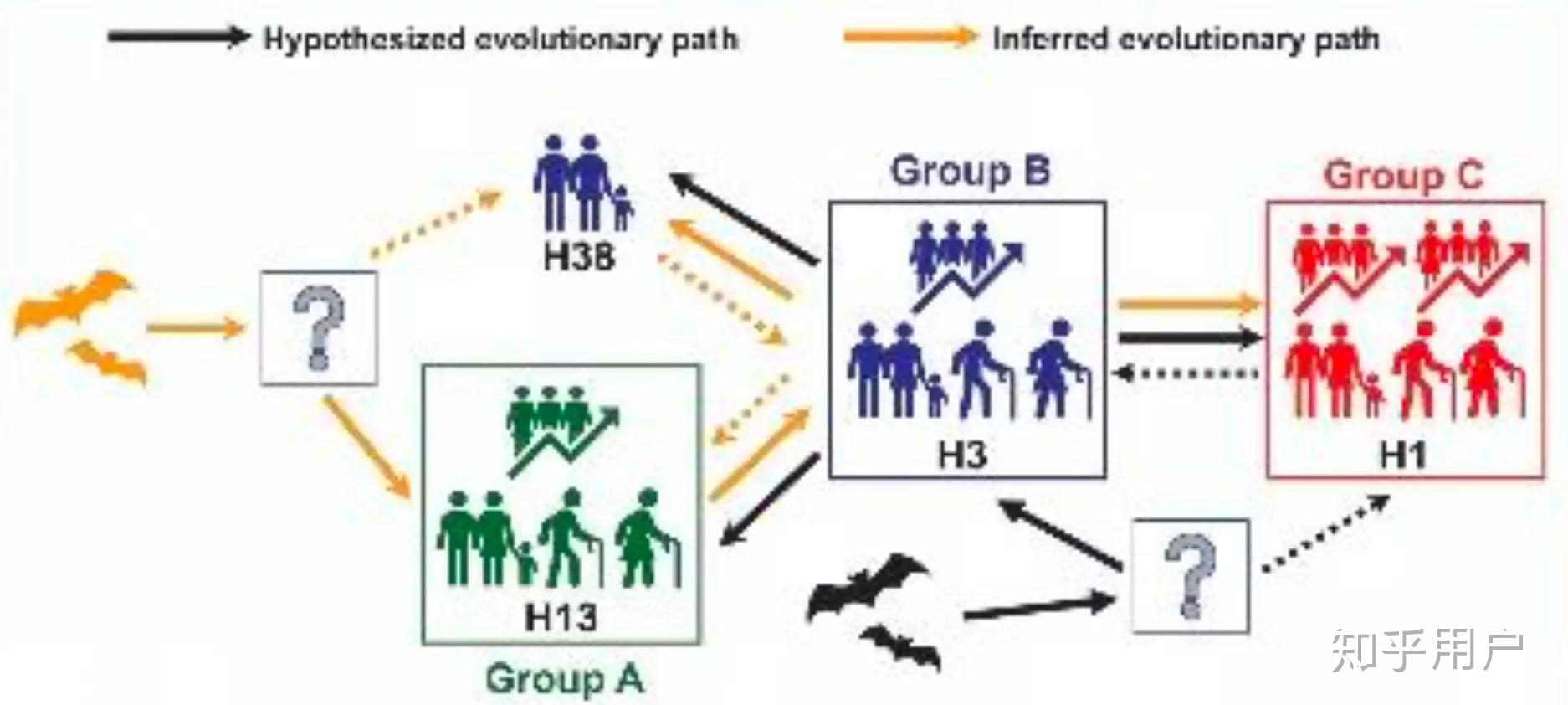
进一步,他们发现,这些单倍型中并不是完全平行,而是存在一个进化上的先后顺序,比如单倍型H3是比较古老的病毒,而H1则是相对较新的病毒。此外,这些病毒的扩散也不是均衡的,比如H1就是一个超级传播单倍型,它衍生出了几十个单倍型。
比较意外的是,他们发现,从武汉华南海鲜市场提取的病毒属于H1类型,就是上图中的下面那个大圈的中心病毒,这个病毒并不是最古老的病毒。从图上大家可以直观的看到,这个H1病毒,并不是很古老的病毒,它源于H3,而H3是H13病毒和H38病毒的后裔,而H38,理论上是蝙蝠病毒RaTG13直系后裔,通过一个未知的中间宿主mv1后形成的。
这意味着,来自华南海鲜市场的病毒并不是最早的病毒,比如更古老的病毒单倍型H13是在广东首例患者-一个来自深圳的病患发现的,而另一个H38病毒则是在美国首例患者——一个来自华盛顿州的病患身上发现。换句话说,这些外地出现的病毒,比华南海鲜市场本身的病毒版本还古老,这意味着,华南海鲜市场并不是最早的源头。
二、其实很早就有人推测华南海鲜市场并不是最早的源头
其实,这个结论并不新鲜,因为,很早的时候,就有研究提到了这个问题。2020年1月24日,顶级医学杂志柳叶刀发表了一篇关于武汉新型冠状病毒疫情的回溯性文章[5],在这篇文章中,我们首次发现了疫情早期来源并不是华南海鲜市场。
最早报道的病例是12月1日,这个病例并没有华南海鲜市场暴露史,接下来12月10日新增的3例患者,有2例无华南海鲜市场暴露史。换句话,在早期爆发的4例中,有3例都是没有华南海鲜市场暴露史的,这就意味着,最早的源头,应该不是华南海鲜市场。
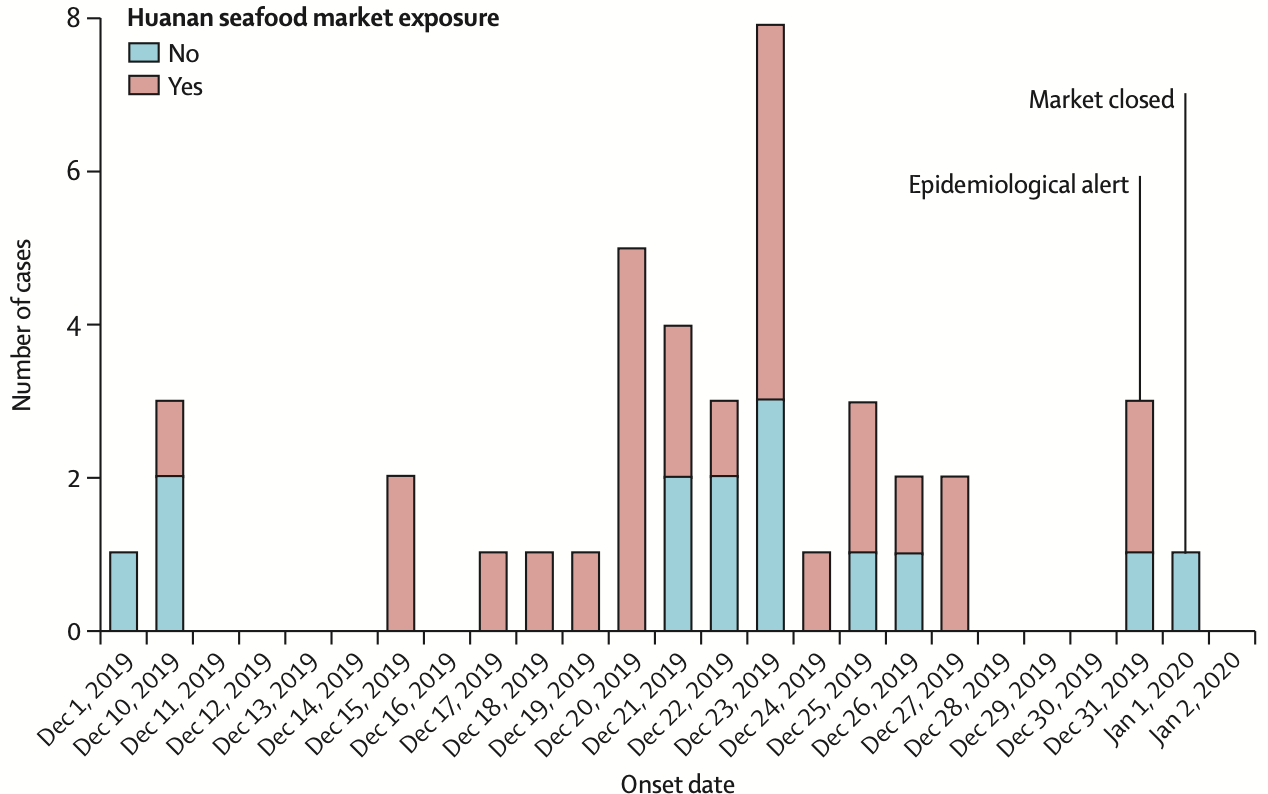
无独有偶,同样另一篇来自新英格兰医学的报道,也佐证了这个事实,如下图所示,浅色的柱状图表示无华南海鲜市场暴露史,而早期病例柱转图,都是浅色的[6]。
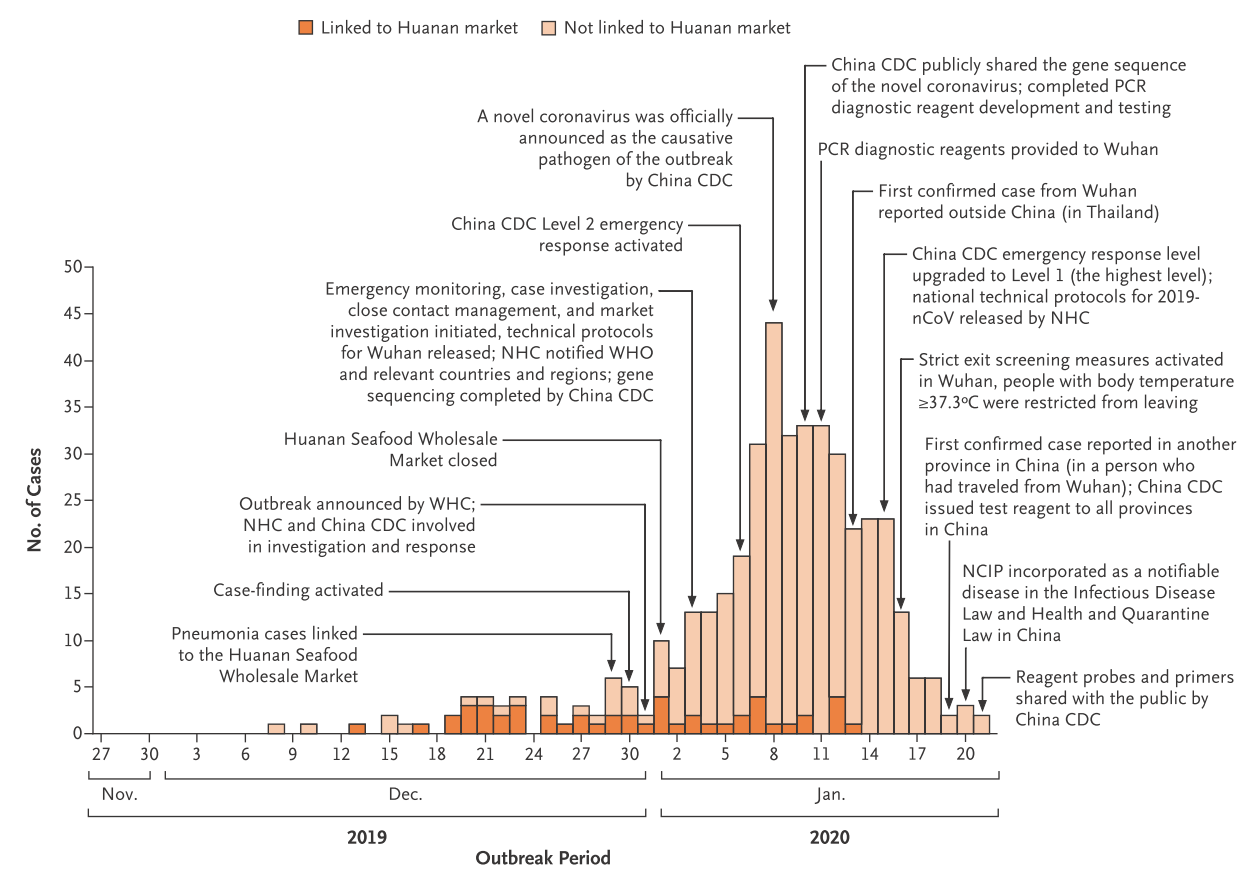
对此,science在1月17日刊发评论,指出,当时对于华南海鲜市场为疫情爆发地点的认识是错误的,疫情应该另有源头[7]。
至此为止,无论是来自病例的溯源,还是来自基因组学的证据,都佐证了一个事实,华南海鲜市场应该不是最早的源头。
1 Chan JF-W, Yuan S, Kok K-H, et al. A familial cluster of pneumonia associated with the 2019 novelcoronavirusindicatingperson-to-person transmission: a study of a family cluster. Lancet 2020
2 HolshueML,DeBoltC,LindquistS,etal.FirstCaseof2019NovelCoronavirusinthe United States. N Engl J Med 2020
3 https://nextstrain.org/ncov
4 WB Yu et al.Decoding the evolution and transmissions of the novel pneumonia coronavirus (SARS-CoV-2) using whole genomic data
5 Chaolin Huang et.al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China, The Lancet, 2020
6 Li Q, Guan X, Wu P, et al. Early transmission dynamics in Wuhan, China, of novel coronavirus–infected pneumonia[J]. New England Journal of Medicine, 2020.
7 Cohen J. Wuhan seafood market may not be source of novel virus spreading globally[J]. Science, 2020.
版权声明:本文系腾讯较真(微信ID:qqjiaozhen)独家约稿,如需转载,请联系较真平台。
较真丨研究发现新冠病毒的源头不是华南海鲜市场,这意味着什么? |